Latest update: 10th Aug. 2015
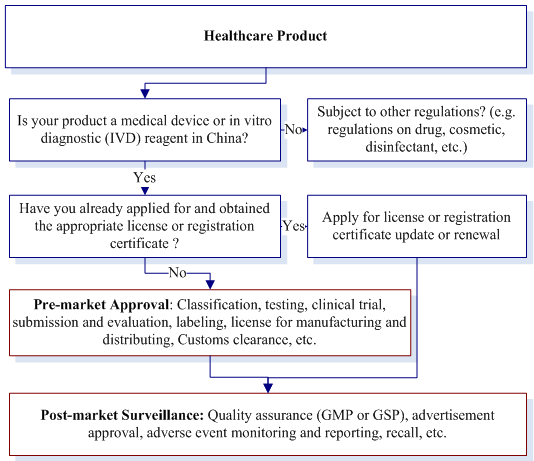
12th Feb. 2014, the revised ‘Regulations for the supervision and administration of medical device’ (revised regulations) has passed by the state council of People’s Republic of China. The Revised Regulations has come into effective on 1 June 2014. The revised regulations reduce the license requirements and adjust the approval duration and administrative department. The class I medical devices are no longer required to register and only required to report for record.
Key contents
- The class I medical devices are no longer required to register and only required to report for record.
- The duration of validity of medical device registration certificate is 5 years.
- The high risk medical devices are required to be approved by CFDA prior to clinical trial in China.
- Legal agent’s information should be indicated on the product instructions.
- The renewal and update of registration certificate are approved separately.
- The registration certificate is the required documents for the manufacturing approval application.
How to Comply
1st Edtion: The Regulations for the Supervision and Administration of Medical Devices (Decree No. 276 2000)
These Regulations are formulated with a view to ensuring the safety and effectiveness of medical device and protecting human health and life safety.
All the medical device activities in the research, manufacture, distributing, use, supervision and administration within the territory of the People's Republic of China shall comply with the Regulation.
Food and drug administration department under the State Council is responsible for supervision and administration of medical devices nationwide. Relevant departments under the state council shall be responsible for supervision and administration related to medical devices, which are within the scope of their respective functions and duties.
Food and drug administration department of the local government at county level and above is responsible for supervision and administration of medical devices in each administrative region. Relevant departments of the local government at county level and above, within the scope of their respective functions and duties, are responsible for supervision and administration related to medical devices.
Food and drug administration department under the State Council shall coordinate with the related departments under the State Council in carrying out and implementing state planning and policies in medical instruments industry.
The state classifies medical devices according to the degree of risk.
Class I Medical devices are those with low risk degree and for which safety and effectiveness can be ensured through routine administration;
Class II Medical devices are those with moderate risk and for which strict administration is required to ensure their safety and effectiveness
Class III Medical devices are those with higher risk and which must be strictly controlled via special measures in respect to safety and effectiveness.
The intended purpose, structure features, use methods and other factors of medical device shall be considered in medical device risk degree evaluation.
Food and drug administration department under the State Council is responsible for making the rules and catalogue of medical device classification and analyzing, evaluating the risk changes of the product in time, adjusting the classification catalogue. The classification catalogue which has been made and adjusted shall be disclosed to the public after asking for comment from medical device manufacturing distributing enterprises, using institution and trade organizations, as well as referring to international medical devices classification practices.
The research and manufacture of medical device shall be based on the principle of safety, effectiveness and saving. The state encourages medical devices research and innovation and will make full use of the market mechanism, promotes the application of medical devices new technology and medical devices industry development.
Medical devices shall comply with the compulsory national standards of medical devices; or shall comply with the compulsory industry standards if there is no compulsory national standard.
Catalogue of disposable medical devices will be made and adjusted by food and drug administration department under the State Council in conjunction with department of national health and family planning competent. Medical devices which can reuse and their safety and effectiveness can be assured will not be listed on the disposable medical devices catalogue. Those shall be adjusted out from the disposable medical devices catalogue if their safety and effectiveness can be assured though the design, manufacturing technique, disinfection technology have been improved.
Medical device industry organization shall enhance self-discipline, promote credibility systems construction, supervise and urge the enterprise to conduct the production and operation activities according to law, guide the enterprise to be honest and trustworthy.
Implement the record management for Class I medical devices, and registration management for Class II, Class III medical devices.
The following materials shall be submitted when record Class I medical devices and register Class II, Class III medical devices:
Applicant for medical devices registration and medical devices recording shall be responsible for the authenticity of the submitted materials.
For class I medical device recording, the applicant shall submit record materials to food and drug administration department of the municipalities consisting of districts.
Test report for class I products can be self-test report of the applicant; clinical evaluation materials for class I shall certify the safety and effectiveness of medical devices and can be achieved based on documents and clinical use data of similar products, while the clinical trials report is not needed.
Class I medical devices manufacturing enterprise abroad which plan to export to china, shall submit the record data and related documents which are approved by the medical device competent departments of overseas governments to prove that the product can enter a certain country (or region) to food and drug administration department under the State Council via its representative offices established within Chinese territory or appointed agent which is the enterprise legal person within Chinese territory.
Make formalities of record amendment when items stated in the record materials have been changed.
The applicant for registering class II medical device shall submit the registration materials to food and drug supervision and management departments of the local provinces, autonomous regions, municipalities directly under the central government. The applicant for registering class III medical device shall submit registration material to food and drug administration department under the State Council.
Class II, class III medical devices manufacturing enterprise abroad which plan to export to china, shall submit the record data and related documents which are approved by the medical device competent departments of overseas governments to prove that the product can enter a certain country (or region) to food and drug administration department under the State Council via its representative offices established within Chinese territory or appointed agent which is the enterprise legal person within Chinese territory.
Test report for Class II, Class III Medical devices shall be presented by medical devices testing and inspection institutes. Clinical evaluation materials for Class II, Class III medical devices shall include clinical trials report, except those which can be exempted clinical trials based on Article 17.
Food and drug administration department which accept the registration shall deliver the application materials to the technical assessment institute within 3 working days after accepting an application. The technical assessment institute shall submit assessment comment to food and drug administration department after finishing technical assessment.
Food and drug administration department which accept the application shall make the decision within 20 working days after finishing the technical evaluation. Permit the applicant to register if the product meets the requirements of the safety and effectiveness, and issue the registration certificate of medical devices; For those do not comply with the requirements, reject the registration and give a written explanation.
Food and drug administration department under the state council shall organize the technical institutions to inspect the quality management system when it is needed during the technical evaluation for imported medical device.
If the design, raw materials, production process, application scope, usage methods, etc. of the registered class II, class III medical devices have been changed substantially, such may affect the safety and effectiveness of medical device, the applicant shall apply for changing the registration to the original registration department. If the change is not substantial and does not affect the safety and effectiveness of medical device, just submit the change to the original registration department for record.
The term of validity for the registration certificate of medical devices is five years. The holder of the certificate shall apply for extending the validity term of registration certificate six months before the certificate expire.
Food and drug administration department which receives extending registration application shall decide before certificate expiration that the product can be registered. If the department fails to make the decision within the specified time, the application is regarded as to be permitted.
A medical device shall not be registered for extending in any of the following circumstances:
The applicant can register the medical devices which are new and have not listed in the catalogue according to the class III registration regulation, and can also determine product categories in accordance with classification principles and apply to food and drug administration department under the state council for category identification, and then do the registration or record procedures based on this regulation.
Food and drug administration department under the state council shall identify the classification of the medical devices which is directly registered as class III according to the degree of risk, and incorporate into the catalogue in time for those permitted to be registered. Food and drug administration department under the state council shall judge the medical devices classification for those which apply for the category identification within 20 working days and inform the applicant.
The clinical trials are not needed when conducting a record for classⅠmedical device, while registering class II, class III medical devices, clinical trials report are required; Clinical trials can be exempted in the following circumstances.
Catalogue for medical device whose clinical trials can be exempted is made, adjusted and issued by food and drug administration department under the state council.
Clinical trials shall comply with the requirements of the quality management criterion for medical device clinical trials and be conducted in the qualified clinical test institution, and submit the record to food and drug administration department of local provinces, autonomous regions or municipalities directly under the central government. Food and drug administration department which accept the record shall publish it to food and drug administration department and the health and family planning competent department where the clinical test institution is located.
The qualification accreditation conditions of clinical test institution and quality management criterion for clinical trials shall be made and issued by food and drug administration department under the state council and the health and family planning competent department. Medical device clinical test institutions are assigned by food and drug administration department under the state council and the health and family planning competent department as well.
Class III medical device clinical trials with higher risk to human body shall be approved by food and drug administration department of the state council. The catalogue of class III medical device clinical trials with higher risk to human body is made, adjusted and issued by food and drug administration department of the state council.
Food and drug administration department under the state council shall analyze the equipments, professionals, risk degree, implementation plan for clinical trials, comparative analysis report of the clinical benefits and risk and other conditions of the assigned clinical test institution comprehensively while approving the clinical trials. The approval shall be informed to the applicant and food and drug administration department and the health and family planning competent department of the local provinces, autonomous regions or municipalities of the clinical test institution.
Enterprises engaged in manufacturing medical devices shall meet the following requirements:
Article 21
Enterprise manufacturing class I medical devices, shall submit the record and certificate materials to food and drug administration department of the municipalities consisting of districts related to the article 20.
Enterprise manufacturing class II and/or class III medical devices, required to apply for manufacturing permission to food and drug regulatory authorities of the provinces, autonomous regions and municipalities directly under the central government and submit proofs related to article 20 and registration certificate for medical device.
Food and drug administration department which accept the application of Medical device Manufacturing Enterprise License shall inspect materials within 30 business days and check based on the requirement of good manufacturing practice of medical device; If meet the requirements, the registration is permitted and Medical device Manufacturing Enterprise License is published to the applicant. For those do not comply with the requirements, reject the registration and give a written explanation.
The term of validity for the registration certificate of medical devices is five years. The holder of the certificate can apply for extending when reach expire according to the administrative license law.
Good Manufacturing Practice for Medical device shall provide explicitly stipulation which may affect the safety and the effectiveness of medical device such as design and exploitation, production equipment conditions, material purchase, production process control, institution setting and personnel allocation of the enterprise.
The medical device manufacturing enterprise shall establish the quality management system related to the medical device according to the requirement of Good Manufacturing Practice for medical device and shall assure the effective operation of medical device quality management system, and organize manufacturing strictly according to product technology which has been registered or recorded, in order to assure the ex-factory medical device to meet compulsory standards and requirements of registered or recorded product technology.
The medical device manufacturing enterprise shall do regular self-examination for the operation of quality management system, and submit the result to the drug regulatory authority of governments of provinces, autonomous regions and municipalities directly under the central government.
The medical device manufacturing enterprise shall implement immediately corrective actions if the production conditions of medical device manufacturing enterprise have been changed and cannot meet the requirements of the medical device quality management system; The manufacture activities shall be ceased immediately and report to the drug regulatory authorities of governments at county level when the safety and effectiveness of medical device are possibly affected.
Medical device shall use common names. The common names shall meet the rules of naming regulation made by food and drug administration department under the state council.
Medical device shall have instruction manuals, labels. The content of them shall be identical with relevant content which is registered or recorded. Medical device instruction manuals, labels shall mark the following items:
The serial number of the Medical device registration certificate and the name, address and contact information of the medical device registrant shall be marked for class II, class III medical devices.
Special explanation of safe use shall be made for medical device which will be used by customers themselves.
The entrusting party is responsible for the entrusted medical device quality. The entrusted party shall meet the requirements stipulated in this regulation for medical device manufacturing enterprise and comply with the corresponding production conditions. The entrusting party shall strengthen management of production behavior of the entrusted party, so that manufacture can meet the legal requirements.
Implantable medical device with high risk cannot be entrusted to a third-party for manufacturing, the detail catalogue will be made and adjusted by food and drug administration department under the state council.
Enterprises distributing medical devices shall have the conditions of possessing appropriate facility(s) and storage conditions for the kind of medical devices to be distributed, possessing medical device quality management system, quality management organization or professional quality management staff;
Enterprise distributing class II medical devices shall submit the record to food and drug administration department of municipality with districts and submit proof materials according to the regulation terms in article 29.
Enterprise distributing class III medical devices shall apply for distributing approval and submit proof materials according to the regulation terms in article 29 to the local food and drug administration department of the government of the municipality consisting of districts.
Food and drug administration department which accept the application shall conduct the investigation within 30 business days since the date of acceptance, and organize the examination if necessary; releasing approval when it meets the regulation conditions and issued Medical device Distribution Enterprise License; When a license is not issued, a written explanation shall be given to the applicant.
The term of validity of the Medical device Distribution Enterprise License is 5 years. The holder of the certificate can apply for extending when reach expire according to the administrative license law.
Enterprises distributing medical devices,users shall inspect qualification of the supplier and medical device qualified certificate when purchasing medical device, and establish a check and inspection record system. Distributing enterprises in wholesaling and class III retails shall establish sales record system.
The record shall included following information:
The check and inspection record system and sales record shall be truthful and kept according to the regulations of food and drug administration department under the state council. The state encourages use advanced technology methods to record.
Transporting and storing medical device shall meet the requirements marked in the instruction manuals, labels; relevant measures shall be taken when special requirements of environment conditions such as temperature and humidity are needed to assure the medical device safety and effectiveness.
Users shall have storage conditions suitable for varieties and quantity of the medical device. The using institution shall strengthen technical training for its staff and operate the medical devices based on the instruction manuals, technical operation specification.
Using institution shall deal with the reusable products according to regulations made by the national health and family planning competent of the People’s Republic of China for disinfection and management.
Disposable medical devices shall not be reused, destroy them in accordance with the relevant provisions of the state and make record.
The users of medical devices shall check, inspect, standardize, maintain the used medical devices according to the requirement of instruction manuals and analyze, evaluate in time to assure they are in good condition and guarantee the quality; build the document system for each large-scale medical device that has long service life, record the content of use, maintaining, transferring and the practical use period. These records shall be kept more than 5 years after the validity termination.
The users shall preserve the original materials for the purchased class Ⅲ products and assure the traceability of the information. In the use of large-scale products, implantable products and intervention products, information such as the name of the product, the crucial technological parameters in conjunction with necessary information related to the safety and quality shall be recorded in medical records or related records.
When discovering the potential danger in medical device, the user shall stop using immediately and inform the manufacture enterprise or other institution which is responsible for the product quality to inspect and repair, the product cannot be used if it cannot reach the safety standards after repair.
Food and drug administration department and health and family planning competent department shall supervise and manage medical device quality and use behavior respectively according to its own responsibility.
Distribution enterprises, users shall not distribute, use medical devices without registration certificates or certificates for qualified products, or medical devices which are past their expiration dates, out of effectiveness, or be eliminated.
Medical device transfer between users,the transferor shall assure the safety and effectiveness of products. Medical devices which are past the expiration date, out of effectiveness, or be eliminated cannot be transferred.
Imported medical devices shall be registered or recorded based on the regulation in chapter 2.
The imported medical devices shall have Chinese instruction manuals and labels. Instruction manuals and labels shall meet the requirements in these rules and relevant compulsory standard, the origin, name, address and contact information of the agents shall be stated clearly in the instruction manuals. Without Chinese instruction manuals and labels or the instruction manuals and labels cannot meet the rules in the regulation cannot be imported.
Entry exit inspection and quarantine institution implement inspection for imported medical devices; medical devices cannot meet the requirements cannot be imported.
Food and drug administration department under the state council shall tell the registration or record situation of imported medical devices to the entry-exit inspection and quarantine departments in China. The entry exit inspection and quarantine institution where the import port is located shall report the imported medical devices clearance situation to food and drug administration department of the government of the municipality consisting of districts.
Enterprises which export medical devices shall assure that the exported medical devices comply with requirements in exported country (region).
Advertisements of medical devices shall be truthful and legal, the content which is false, exaggerated or misleading are forbidden. Advertisements of medical devices shall be reviewed and approved by food and drug administration department under the local people's government of the province, autonomous region or municipality and the medical devices manufacturing enterprise or agents of imported medical devices can obtain documents of medical devices advertisement approval. The advertiser shall inspect and check the documents of advertisement approval and its reality before advertising. The advertisement content cannot be issued without advertisement approval or the reality of the approval has not been checked or the content does not identify with the approval. Food and drug administration department of provincial, autonomous regional and municipal people's governments shall publish and update the approved medical devices advertisement catalogue and content in time.
Medical devices advertisement cannot be published in suspension period, which are ordered by food and drug administration department above the provincial level to cease the manufacture, sale, import and use. Medical devices advertisements review and approval provisions are made by food and drug administration department under the state council and administration for industry & commerce under the state council
The state shall establish a medical device adverse event supervision system to collect, analyze, evaluate and control the medical device adverse event.
Enterprises manufacturing, distributing or using medical devices shall conduct the supervision of adverse event and report the adverse event or doubtful adverse event to the medical devices adverse event monitoring technical institutions according to the regulations of food and drug administration department under the state council.
Any institution or individual has the authority to report the adverse event or doubtful adverse event to food and drug administration department or medical devices adverse event monitoring technical institutions.
Food and drug administration department under the State Council shall strengthen construction of network information for monitoring adverse event. Medical device adverse event monitoring technical institutions shall strengthen adverse event information supervision, collect adverse event information actively; when discovering or accepting the report of adverse event, shall check, investigate, analyze and evaluate the adverse event timely, and propose treatment suggestion to food and drug administration department and the health and family planning competent department
Medical devices adverse event monitoring technical institutions shall promulgate the contact information to manufacturing and distributing enterprise,using and consuming institution to report the adverse event of medical device.
Food and drug administration department shall promulgate warning-information, order to suspend manufacture, sale, import and use or other control measures according to the evaluation results of adverse event timely. Food and drug administration department of people's government at or above the provincial level and the health and family planning competent department or relevant departments at the same level shall organize investigation and treatment timely for medical devices adverse event which is outburst, or with large population or have cause serious injury or death, and supervision of similar which caused serious injury shall be strengthened as well.
Manufacture and distribution enterprise and using medical devices shall coordinate with the investigation which conducted by medical devices adverse event monitoring technical institutions and food and drug administration department.
Food and drug administration department of people's government at or above the provincial level shall reassessment the registered medical devices that have following circumstance:
When the consequences of the reassessment for registered medical devices cannot ensure the safety and effectiveness, the original issuing department shall cancel registration certificate of medical devices and publish it to the society. Manufacture, import, sale and use of the medical device are forbidden when the registration certificate is canceled.
Enterprises manufacturing medical devices shall cease production when discovering the medical devices cannot comply with the compulsory standard, the registered or recorded technical requirements or other defect, and inform the related distribution enterprises, users and consumers to stop distributing or using, and recall the medical devices which are working on the market and take relevant measures to remedy and destroy, etc.. Recording and publishing relevant information, report the recall and treatment to food and drug administration department and the health and family planning competent department.
Enterprise distributing medical devices shall stop operation and inform the related manufacture and distribution enterprises, the users and consumers when discovering the medical devices that are in the preceding paragraph’s condition. Enterprises manufacturing medical devices shall recall the medical devices that are in the preceding paragraph’s condition. Food and drug administration department will order manufacture and distribution enterprises to recall and stop operation if they violate the rules.
Food and drug administration department shall strengthen the supervision and inspection of medical devices registration, recording, manufacturing, distribution and use activities. Following events shall be supervised and inspected significantly.
Article 54
Food and drug administration departments have the following authorities:
Food and drug administration departments shall show their law enforcement certificates when performing their duties and keep the commercial secrets of the institution under inspection.
The relevant institution and individuals shall cooperate in the supervision and inspection and cannot hide the relevant information.
Food and drug administration departments shall take urgency control measures such as suspending production, import, sale or use for medical devices which endanger or have proofs which can certify the danger to human health.
Food and drug administration departments shall strengthen sampling inspection for medical devices manufactured, operated or used in the related institution and cannot ask for inspection fees and any other fees, the needed fees will be paid via government budget at the corresponding levels.
Food and drug administration department above the provincial level of people’s government shall publish the quality announcements based on the results of sampling inspection.
Quality accreditation work of medical devices inspection institution shall be unified control according to relevant regulations of the state. The inspection institution shall be identified by certification and accreditation supervision department and food and drug administration department under the state council before implementing inspection.
Food and drug administration department shall entrust the qualified medical devices testing and inspect institution and pay for the relevant fees when the inspection of medical devices is needed in enforcing the law.
The parties can choose a qualified medical devices testing and inspection institution and ask for re-inspection within 7 business days since receiving the inspection results if diagree with it. The medical devices testing and inspection institution which offer the re-inspection shall give the re-inspection conclusion within the time period stipulated by food and drug administration department under the state council. The re-inspection result is the final conclusion.
The medical devices may be with hazardous substances or the design, materials and production processes are changed and have potential risk, medical devices testing and inspection institution can supplement test items and methods for inspection if the test items and methods in national standards and industry standards cannot be inspected. The inspection conclusions achieved in the supplemental test items and methods can be the basis for the quality of medical devices after approved by food and drug administration department under the state council.
The government of the municipality consisting of districts and county government food and drug administration departments shall strengthen supervision and inspection of medical devices advertising; Report the advertisement which is without permission or its content has been tampered to provinces, autonomous regions and municipalities food and Drug administration department, and announce to the comminstitutiony.
Administrations for industry and commerce departments shall supervise and inspect of medical device advertising, penalize illegal acts according to laws and other administrative laws and regulations relevant to advertisement.
Food and drug administration department under the State Council shall establish information platform for supervision and administration of medical devices. Food and drug administration department shall publish regular supervision and management information such as administrative licensing, recording, inspection, illegal activities investigation, etc. via information platform. However, the parties involved shall keep the business secrets.
Food and drug administration departments shall establish credit system for persons who register or record the medical device, production and operation of enterprises and the user; increase the supervision and inspection frequency to a bad credit record institution according to the credit records.
Food and Drug Administration departments and other institution shall publish its contact information, accept counseling, complaints and reports. Food and Drug Administration departments and other institution shall reply in time when received counseling; shall verify and handle when receiving complaints and reports; shall record and store the circumstances of reply, verification and treatment for the counseling, complaints and reports.
If the report for medical device development, production, operation, usage behavior is substantiated after investigation, food and drug administration departments shall reward whistleblower.
The food and drug administration departments under state council shall make and adjust the catalogue stipulated in this regulations and related standards for medical devices supervision and management, shall ask for comments in public as well; The gathering of opinions may be in various forms such as panel discussion, feasibility study meeting, in order to gather comments from medical equipment manufacturing, distributing enterprises as well as users, consumers and other aspects.
If has the following circumstances, food and drug administration departments at county level and above shall confiscate the illegal income, illegal manufactured medical devices and tools, equipments, raw materials used in production and operation. If monetary value of the illegal medical devices is less than 10000 yuan, the violator shall be fined not less than 50000 yuan but not more than 100000 yuan concurrently. In case the monetary value exceeds 10000 yuan, a fine of 10 to 20 times of the monetary value; In serious cases, permit application applied by responsible people and enterprise will be refused within 5 years.
When occurring the preceding paragraph condition and with serious consequence, the Manufacturing Enterprise License or Medical Device Distributing Enterprise License shall be canceled by the authorities which originally issued the license;
Achieve Registration Certificate for Medical Device, Medical Device Manufacturing Enterprise License, Medical Device Distributing Enterprise License,approval documents for advertisement and other approved certificates through false materials or other deception means, the authorities which originally issued the license shall repeal the achieved license and require less than 50000 yuan penalty but not more than 100000 yuan. Responsible people and enterprise will not be allowed to apply for registration of medical device within 5 years.
The authorities which originally issued the certificates shall revoke or confiscate the certificates which is forged, changed, purchased or sold, rented, lent and confiscate the illegal income, if illegal income is less than 10000 yuan, the violator shall be required to pay less than 10000 yuan but not more than 30000 yuan pernalty. In case the illegal income exceed 10000 yuan, a pernalty of 3 to 5 times of the total sum of illegal income; Constitutes a violation of public security administration, the public security organ shall punish it according to the public security management.
Food and drug administration departments at county level and above shall order those do not record according to this regulation to make corrections within the specified time period. The enterprises and products do not correct within the time will be published to the society and receive less than 10000 yuan penalty.
When fake materials are submitted for the record, the related enterprise and the name of the product will be published to the society by food and drug administration departments at county level and above. In serious cases, directly responsible persons cannot be engaged in manufacturing, distributing medical devices activities within 5 years.
If has the following circumstances, food and drug administration departments at county level and above shall order to make corrections and confiscate relevant medical devices in manufacturing, distributing and usage links; If monetary value of the illegal medical devices is less than 10000 yuan, the violator shall be fined not less than 20000 yuan but not more than 50000 yuan concurrently. In case the monetary value exceed 10000 yuan, a fine of 5 to 10 times of the monetary value; In serious cases, order of suspension of production or business even the Registration Certificate for Medical device, Manufacturing Enterprise License and Medical device Distributing Enterprise License may be revoked by the authorities which originally issued the license.
Article 67
If has the following circumstances, food and drug administration departments at county level and above shall order to make corrections and the violator shall be fined not less than 10000 yuan but not more than 30000 yuan concurrently; In serious cases, order of suspension of production or business even the Manufacturing Enterprise License and Medical device Distributing Enterprise License may be revoked by the authorities which originally issued the license.
Article 68
When following circumstances occur, food and drug administration departments and the health and family planning competent department at county level and above shall order to make corrections and give warnings based on its own duties; If refuse to correct, the violator shall be fined not less than 5000 yuan but not more than 20000 yuan concurrently; In serious cases, order of suspension of production or business even the Manufacturing Enterprise License and Medical device Distributing Enterprise License may be revoked by the authorities which originally issued the license.
Article 69
If carry out clinical trials of medical devices that violate this Regulation, food and drug administration departments at county level and above shall order to make corrections or suspend immediately, and the violator may be fined less than 50000 yuan; In serious case, the persons who are directly in charge and the other persons who are directly responsible for the clinical institutes shall be degraded,dismissed or fired according to the law;Besides, the qualification of clinical trials institution would be repealed by the authorities that originally issued the qualification, and qualification recognition application will be refused within 5 years;
If clinical trials institution and its staffs issue false reports,its qualification will be repealed by the authorities which originally issued the qualification, and qualification recognition application will be refused within 10 years; Food and drug administration departments at county level and above can fine the violator less than 50000 yuan but not more than 100000 yuan; Confiscate the illegal income if has; The persons who are directly in charge and the other persons who are directly responsible for the clinical institutes shall be dismissed or fired according to the law;
If medical devices testing and inspection institute and its staffs issue false reports,its qualification will be repealed by the authorities which originally issued the qualification, and qualification recognition application will be refused within 10 years; fine the violator less than 50000 yuan but not more than 100000 yuan; Confiscate the illegal income if has; The persons who are directly in charge and the other persons who are directly responsible for the institutes shall be dismissed or fired according to the law;Persons who are fired cannot engaged in medical devices testing and inspecting work within 10 years from the date of making the decision.
If the issued advertisement violate the provisions of this Regulation,or the advertisement is without approval or the reality of the approval has not been checked or the content does not identify with the approval, the industrial and commercial authority shall punish it according to relevant laws and regulations for advertisements management.
If the content of advertisement has been tampered, approval documents for advertisement will be repealed by the authorities which originally issued the documents,and such approval documents recognition application will be refused within 2 years.
Food and drug administration department above the provincial level can decide to suspend selling the medical devices with false advertising, the decision will be published to the society; If still sale this kind of medical devices, food and drug administration departments at county level and above shall confiscate the illegal medical devices and fine less than 20000 yuan but not more than 50000 yuan;
The technical assessment institute, medical devices adverse event monitoring technical institutions do not conduct work according to the regulation in this Regulation, or are serious irresponsible and caused major mistake in the assessment and supervision, food and drug administration departments at county level and above shall order to make corrections, circulate a notice of criticism, give warnings; If caused serious consequences, the persons who are directly in charge and the other persons who are directly responsible for the clinical institutes shall be degraded,dismissed or fired according to the law;
Food and drug supervision and administration department and its staff shall exercise the power of administrative sanctions strictly in accordance with the regulations of categories of the penalties and extent based on property of illegal behavior and concrete details.
If food and drug administration departments at county level and above or other departments do not perform regulatory responsibilities, derelict duty or engage in favoritism, or other acts which violate the regulation in this Regulation, the persons who are directly in charge and the other persons who are directly responsible for it will be given sanctions such as warning, recording a demerit, or recording a serious demerit by supervisory organ or appointment and removal institution; If has caused serious consequences, the persons who are directly in charge and the other persons who are directly responsible for shall be degraded,dismissed or fired according to the law.
In violation of provisions of these Regulations, to the extent to which crimes are committed, criminal liabilities shall be investigated and handled according to the law; For those caused damage to person or property or other damages, shall be liable for compensation according to law.
Meaning of the following terms in the Regulations:
"Medical devices" as defined by this Regulation refers to: any instrument, apparatus, appliance, material, in vitro diagnostic reagents and calibration substances and other similar substances and related articles, including the needed computer software. Its main effectiveness is achieved via physics ways and so on. It does not achieve its principal action in or on the human body by means of pharmacology, immunology or metabolism, but which may be assisted in its function by such means; the use of which is to achieve the following intended objectives:
Medical device users, refers to organization which use medical device to provide medical and other technical services for others, including medical institution which has obtained “Practice License of Medical Institution” and family planning technical services institution which has obtained” “Practice License of family planning technical services” and blood bank, blood plasma stations, rehabilitation aids assembly institution which do not need to obtain “Practice License of Medical Institution”.
Medical device registration fee may be charged. The fees for specific items and charging standards shall be made by the responsible finance and price department of the state council in accordance with relevant state regulations.
Provisions governing non-profitable contraceptive devices and devices responsible for public health emergencies, shall be formulated by food and drug administration departments in conjunction with the health and family planning competent department under the State Council according to this Regulation;
Provisions for Chinese medical device management shall be formulated by food and drug administration departments under the State Council in conjunction with the state Administration of Traditional Chinese Medicine according to this Regulation; Provisions for the scope of rehabilitation aids and its management shall be formulated by food and drug administration departments in conjunction with the civil affairs department under the state council according to this Regulation.
Specific provisions for supervision and management of military medical devices shall be organized and implemented by military health administrative department based on this Regulation and related regulations in military.
These Regulations shall come into force from June 1, 2014.